Cleft palate and cleft lip surgery is one of the main reasons why cosmetic surgery exists. This affects 1 in 700 children all over the world.
At Marina Plastic Surgery, our top surgeons have devoted their lives to bringing smiles to children all over the world who suffer from cleft lip & palate abnormalities. This level of commitment is what has given us the highest level of expertise in our chosen field.
Aside from the aesthetic value of having an operation done to treat this congenital abnormality, it really improves the patient’s quality of life. We also treat other forms of congenital anomalies to improve the lives of children all over the world.
Overview
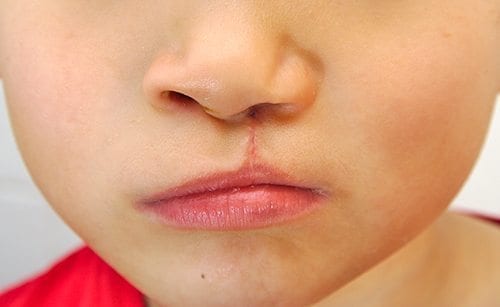 There are many forms of congenital anomalies that affect people all over the world. Here is a list of congenital anomalies we have observed and operated on. Our surgeons are highly qualified to diagnose and offer advice or treatment as to what to do next with your unique case.
There are many forms of congenital anomalies that affect people all over the world. Here is a list of congenital anomalies we have observed and operated on. Our surgeons are highly qualified to diagnose and offer advice or treatment as to what to do next with your unique case.
Best Candidates
Young children 3 to 6 months or up to 3 years old with any of the congenital anomaly listed above with good general health and as close as possible to their ideal BMI based on age, height and other physical factors.

Martin Rees
MB ChB. FRACS
Martin Rees is a Cosmetic and Reconstructive Plastic Surgeon who has been in private practice since 1988, founding the Auckland…
Craniosynostosis
Craniosynostosis is the condition wherein the fibrous joints between the cranial sutures fuse prematurely. This hampers normal brain and skull growth producing an asymmetrical appearance. Beyond the aesthetic effect of craniosynostosis, the compressed space can cause unnecessary pressure inside the skull.
There are 4 types of craniosynostosis:
Saggital – premature fusion of the saggital suture that runs from the front to the back at the top of the skull.
Coronal – premature fusion of the suture running from ear to ear at the top of the skull.
Metopic – this suture runs from the top of the bridge of the nose to the midlineof the forehead and connects to the saggital suture. Also called trigonocephaly.
Lambdoid – the rarest form of craniosyntosis that makes the head appear flat on one side with one ear higher than the other or a tilted appearance of the head to one side.
This is a serious condition that can lead to visual impairment, sleep disorder, mental development impairment leading to a severely reduced IQ.
The causes for this condition have yet to be fully determined although genetics play a huge part in the development of this anomaly. Other causes are environmental or hormonal. Smoking, ingestion of amine-containing drugs are highly suspected factors.
Here are other related cephalic disorders:
Scaphocephaly ( Canoe-shaped skull)
This condition results from premature fusion, or inadequate growth along the midline or sagittal suture, which runs from front to back along the top of the skull. Because there is no side to side growth, these skulls are long and narrow.
This condition can be corrected by surgery which is also highly advised to relieve intracranial pressure. The use of a cranial remoulding orthosis can correct the condition if detected early and used immediately.
Brachycephaly (Short skull)
This condition is caused by premature fusion or inadequate growth across the coronal sutures, which run from the “soft spot” on the top of the baby’s head, down into the temples on either side. In Classic brachycephaly, both left and right coronal sutures are involved.
This condition can be corrected by surgery which is also highly advised to relieve intracranial pressure. The use of a cranial remoulding orthosis can correct the condition if detected early and used immediately.
Plagiocephaly (Oblique Head)
This characteristic deformity is quite common nowadays, due to infants being placed on their backs to sleep. This trend started to prevent crib death in infants. The problem is that newborn skulls are soft and still malleable in the early stages making it easily deformed if kept in one position. Gravity plays a part in this, externally applied pressure can also contribute to the development of an oblique head.
This can be corrected with the use of a cranial remoulding orthosis.
Trigonocephaly (Pointed, triangular head)
This is thought to be due to premature fusion of a midline suture in the frontonasal/forehead region. It produces inadequate growth across the forehead and hence a characteristic triangular deformity of the front of the head. Again, this is treated by Craniofacial surgical intervention.
As with all other cases of premature or incorrect fusion, this can be corrected with surgery or the use of a cranial remoulding orthosis.
Crouzon’s Disease
This is a genetic disorder which causes the premature fusion of cranial sutures. This severely affects the shape of the head and the face.
Strip craniectomy or open vault surgery is used to prevent premature fusion of cranial sutures. This allows the brain to normally develop within the given time period.
A cranial remoulding orthosis may be used afterwards.
Apert’s Syndrome
Apert’s syndrome affects not just the head but the hands and feet as well. Patients with Apert’s syndrome are characterized as having a brachycephalic head with syndactyly present in hands and feet. Dental issues may occur due to the malformation of the skull. This is caused by premature fusion or craniosynostosis of the skull. This has other effects including reduced IQ, hampered brain development, asymmetrical facial features, dental problems, clefting and others.
Surgery can be used to correct the craniosynostosis while additional surgery to the hands and feet can correct the fusion of the extremities.
Binder’s Syndrome (Nasomaxillary Dysplasia)
The cause of this congenital anomaly has yet to be determined. Patients affected by Binder’s syndrome are often characterised as having a flattened midface appearance with a noticeable sunken bridge of the nose or a severely flattened appearance. The jaw may protrude and have a pronounced overbite. This results in difficulty of breathing, snoring, nasal infections, speech impediments, dental issues and others.
Surgery is recommended at an early age to immediately correct the condition.
Hemi-facial or Craniofacial Microsomia ( Lateral facial) dysplasia
This congenital anomaly affects the development of the face especially the lower half, ears, mouth and jaw. It may affect one or both sides of the face.
Blood clotting or insufficient blood supply is the suspected cause for this condition during the gestation period. This can also be caused by physical trauma or genetics. The definitive reason why this happens has yet to be confirmed.
Surgery to correct the deformity is the best course of action. Distraction osteogenesis can slowly repair this condition through a series of breaking the bones and allowing it to heal to fill the gap. A graft might be used to repair the lower jaw.
This congenital anomaly shares a lot of features/characteristics as the Treacher-Collins Syndrome.
Treacher-collins Syndrome
The Treacher-Collins Syndrome is a congenital anomaly that happens to 1 out of every 50,000 people. The severity of the case varies from one patient to the next from very mild to severe. Complications arising from this include breathing disorders, hearing loss, cleft palate and vision problems. It can also affect the person’s IQ as most patients with Treacher Collins Syndrome are of average intelligence.
The most common symptom of Treacher Collins Syndrome is an underdeveloped jaw and lack of cheek bones. Due to this, eyes may appear slanting downwards. Affected patients may also have very small to non-existent ears. Patients may suffer from breathing problems, speech disorders, lack of vision, hearing loss.
This condition can be corrected through a series of cosmetic surgery operations.
Congenital Hand Anomalies
Hands are also prone to deformities, derangements or underdevelopment.
Polydactyly
Polydactyly is caused by a mutation in the gene resulting in an excess of digits in the hands, feet or both. This can be corrected through surgery.
Syndactyly
Syndactyly is the condition wherein digits are fused together due to a lack of separation during the fetus’ development in the womb. Separation of the digits can be done depending on the severity of the condition with the use of skin grafts to close off the operated portions.
What to Expect
Dealing with congenital anomalies is a sensitive matter.
The main goal of congenital anomaly correction surgery is to improve the patient’s appearance and quality of life.
An initial consultation will be done to assess what you essentially need for functionality with a secondary assessment done for the aesthetic purpose of the operation.
Young children with any of the congenital anomaly listed above with good general health and as close as possible to their ideal BMI based on age, height and other physical factors.
There may be damage to deeper structures not immediately evident during the initial consultation. Additional surgical procedure may need to be done to fully correct severe cases.
After Surgery / Recovery
As with all types of surgical procedures, caring for the areas where sutures were made is the highest priority. Follow your surgeon’s instruction for the proper care of affected areas.
Utmost care and priority must be given to ensure the safety of the individual’s operated areas during the recovery period.
Scarring may remain slightly pink for six months or more before they fade to a thin, barely visible line.
Younger children may require arm splints (sometimes called “no-nos”) to prevent accidental scratching/touching/impact on operated areas during the course of the day.
A cranial remoulding orthosis may be required for patients needing skull correction operations. These helmets are custom built for each individual and can be designed to look more aesthetically pleasing with bright colours or graphics.
Procedure Planner
Initial Meeting
45 – 60 minutes
Pre-op
30 – 45 minutes
Procedure Time
2 – 6 hours (procedure time is highly dependent on congenital anomaly presented)
Recovery Time
3 – 4 weeks
Post-op Follow Up
1 week